Blogbeitrag: Klinische Bewertung
Hersteller verbringen viel Zeit mit der Diskussion über die Umsetzung von MDR und MDCG Leitlinien. In den Unternehmen, mit Benannten Stellen und auf Austauschveranstaltungen. Bei der klinischen Bewertung scheint oft das Ziel in den Hintergrund zu rutschen. Uneinigkeit bei Interpretationen der Regularien in Kombination mit Halbwissen durch Mundpropaganda und Streit über Formalitäten führen zu einem toxischen Cocktail. Dieser verschwendet Ressourcen und macht Medizinprodukte weder besser noch sicherer. Lasst uns den Blick wieder auf das Wesentliche richten.
Inhalt dieses Blogbeitrags:
- Zweck und Ziel der klinischen Bewertung
- Fokus bei der Durchführung
- Unsinn bei Dokumentation und Prüfung von klinischen Bewertungen
- Den Unsinn beseitigen: Forderungen an Hersteller und Benannte Stellen
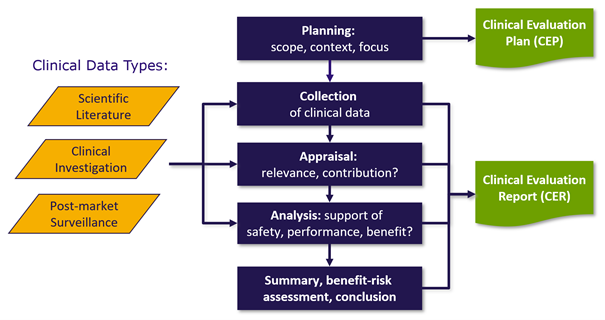

Die gleichen regulatorischen Spielregeln aus der MDR, ergänzt durch Europäische Leitlinien, müssen für einen extrem inhomogenen Markt mit unterschiedlich Rahmenbedingungen angewendet werden. Dessen war sich der Gesetzgeber bewusst (MDR, Artikel 61):
„Der Hersteller spezifiziert und begründet den Umfang des klinischen Nachweises […]. Der Umfang […] muss den Merkmalen des Produkts und seiner Zweckbestimmung angemessen sein.“
Bedeutet im Klartext:
Der Hersteller muss sich Gedanken machen, welche Daten für den Nachweis der jeweils produktspezifischen Sicherheits-, Leistungs- und Nutzenaspekten herangezogen werden sollen und (fundiert!) begründen, warum er denkt, dass diese am Ende ausreichen (= klinische Strategie).
Das ist die Aufgabe des Clinical Evaluation Plan (CEP). Dort wird das geplante Vorgehen festgelegt, begründet und kann bei Bedarf frühzeitig von der Benannten Stelle geprüft werden. Das ist vor allem wichtig, wenn Sie eine klinische Prüfung durchführen müssen, denn dann sollte das Ziel der Studie zur klinischen Strategie im CEP passen.
Der Clinical Evaluation Report (CER) fasst am Ende der Entwicklung, während der Produktvalidierung, die Ergebnisse dieser Aktivitäten zusammen, diskutiert und gleicht sie mit dem Risikomanagement Report ab. Es folgt die Schlussfolgerung, ob die Ziele im CEP auch erfüllt wurden.
Einfach formuliert:
„Im CEP beschreibe ich produktspezifische Fragen.
Im CER weise ich nach, dass ich diese Fragen basierend
auf klinischen Daten beantworten konnte.“
Seit 2011 führe ich Seminare zum Thema klinische Bewertungen durch, habe viele Jahre selbst klinische Bewertungen geschrieben und Hersteller bei der Durchführung beraten und unterstützt.
Was kein Seminarteilnehmer gerne hört, aber Realität ist: Klinische Bewertungen sind individuell. Produkthistorie, Innovationsgrad, vorhandene Input Dokumente, relevante Sicherheits-, Leistungs- und Nutzenaspekte, die Wahl von Datenarten, identifizierte Daten, Diskussion der Ergebnisse, usw. Deshalb nehmen Erstellung und Prüfung der klinischen Bewertung viel Zeit in Anspruch und auf beiden Seiten müssen Experten mit entsprechend technischem und medizinischem Fachwissen beteiligt sein (Sie würden bei einer Augenentzündung auch nicht zum Urologen gehen, obwohl beide Ärzte sind).
Diskussionen zwischen Hersteller und Benannter Stelle lassen sich per se kaum vermeiden, da unterschiedliche Expertise, Sichtweisen, Erfahrungen und Erwartungshaltungen aufeinandertreffen.
Was mich zunehmend irritiert:
„Viele Diskussionen zwischen Hersteller und Benannter Stelle sind nicht mehr zielführend und die Verunsicherung der Hersteller sowie Berater/Dienstleister steigt.“
Das führt zum einen dazu, dass CEP und CER immer länger werden. Es wird möglichst viel Text erzeugt in der Hoffnung, bloß keine Abweichungen zu kassieren.
Wie würden Sie sich fühlen, wenn Sie sich durch mehrere hundert Seiten wühlen müssten, um irgendwo die Quintessenz der klinischen Bewertung (Antworten auf bestimmte Fragen) zu finden? Und dann stolpern Sie auch noch über triviale Fehler wie Inkonsistenzen (der Zweckbestimmung, Risiken, Marketing Claims…)? Willkommen im Leben der Clinical Reviewer.
„The more the better“ ist KEINE Lösung.“
Ein weiterer Aspekt sind Berichte von Abweichungen, die keine regulatorische Grundlage haben:
- "Die klinische Bewertung muss in allen Landessprachen vorhanden sein."
- "Ein medizinischer Experte muss mindestens 5 Publikationen pro Jahr veröffentlichen, sonst ist er nicht als Reviewer geeignet."
- "Im CER darf nur eine Art von klinischen Daten verwendet werden: Entweder Literatur oder klinische Prüfung. Entscheiden Sie sich."
- "Es dürfen nur Daten von einem einzigen Equivalent Device herangezogen werden."
- "Die klinische Bewertung muss aus einem Einzeldokument bestehen, in dem alle Informationen zu finden sein müssen. Referenzen werden nicht akzeptiert."
Aber auch Hersteller bekleckern sich nicht mit Ruhm. Viele Abweichungen und daraus resultierende Nacharbeiten können einfach vermieden werden. Es fehlt Wissen über regulatorische Anforderungen oder ein Verständnis für deren praktische Umsetzung. Manchmal werden Inhalte ohne sinnvollen roten Faden und Fokus auf das Ziel zusammengestellt. Auch Inkonsistenzen sind ein typisches Problem, welche durch vielfaches Copy & Paste und spätere Änderungen von Information entstehen (was im Übrigen mit digitalen Lösungen heutzutage einfach behoben werden kann).
In Summe: Viel Text, Zeit und Geld - aber leider oft ohne Mehrwert.
„Zielführender: Den Fokus beim Erstellen von CEP und CER auf relevante „Fragen und Antworten“ legen.“
Alle Stakeholder unserer Branche sitzen eigentlich in einem Boot: Wir kämpfen mit teils schlecht geschriebenen Regularien und müssen einen praktikablen Weg für deren Umsetzung finden. Dabei könnte ein Perspektivenwechsel und der Fokus auf die eigentliche Aufgabe helfen: Sichere und erfolgreiche Medizinprodukte mit einem nachgewiesenen Nutzen auf den Markt bringen.
Solange weiterhin über Nichtigkeiten wie der "wahren" Definition von Intended Purpose und Intended Use gestritten wird, verschwenden wir alle Zeit und Geld mit Belanglosigkeiten – jeden Tag.
Muss das sein? Wir haben die Wahl!
Ihr habt Anmerkungen zum Blog oder möchtet mehr über dieses Thema erfahren?
Dann freuen wir uns über Ihre Kontaktaufnahme.
